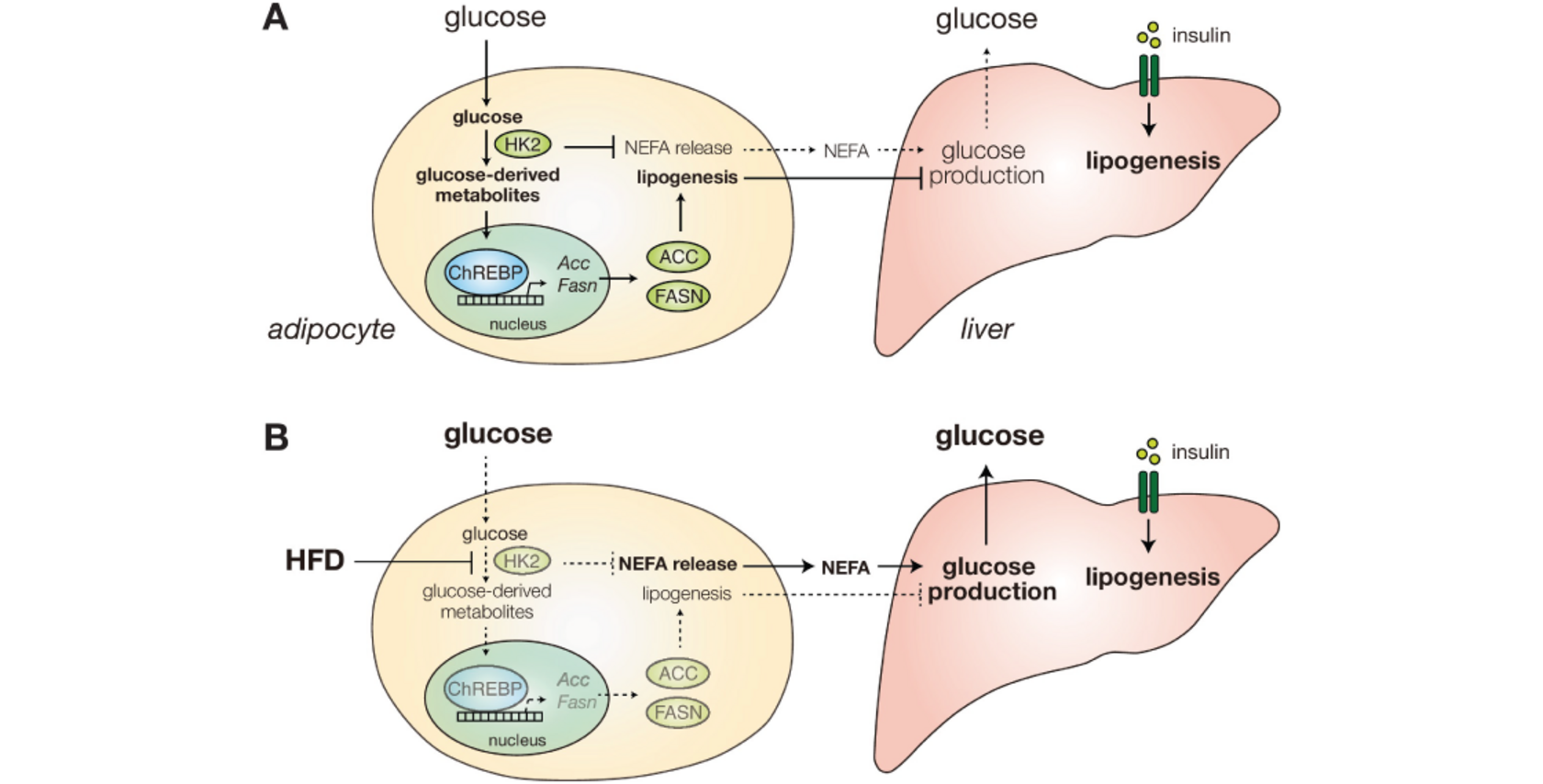
Diabetes and fatty liver disease can be caused by hyperglycemia which in turn can be caused by obesity. However, the linking mechanism is unkown. The lab of Michael Hall and collaborators could now show that a high-fat diet in mice leads to the loss of Hexokinase 2 in adipose tissue. This loss occurs through translation inhibition of translation of the mRNA encoding Hexokinase 2. Interestingly, Mexican cavefish, which are hyperglycemic, have a loss of function mutation in the gene encoding Hexokinase 2. In summary, the obesity induced loss of Hexokinase 2 seems to be a key step in the development of hyperglycemia and diabetes, and this mechanism seems to be evolutionarily conserved. Their findings were published in the article "Diet-induced loss of adipose hexokinase 2 correlates with hyperglycemia" in Elife.
Abstract
Chronically high blood glucose (hyperglycemia) leads to diabetes and fatty liver disease. Obesity is a major risk factor for hyperglycemia, but the underlying mechanism is unknown. Here, we show that a high-fat diet (HFD) in mice causes early loss of expression of the glycolytic enzyme Hexokinase 2 (HK2) specifically in adipose tissue. Adipose-specific knockout of Hk2 reduced glucose disposal and lipogenesis and enhanced fatty acid release in adipose tissue. In a non-cell-autonomous manner, Hk2 knockout also promoted glucose production in liver. Furthermore, we observed reduced hexokinase activity in adipose tissue of obese and diabetic patients, and identified a loss-of-function mutation in the hk2 gene of naturally hyperglycemic Mexican cavefish. Mechanistically, HFD in mice led to loss of HK2 by inhibiting translation of Hk2 mRNA. Our findings identify adipose HK2 as a critical mediator of local and systemic glucose homeostasis, and suggest that obesity-induced loss of adipose HK2 is an evolutionarily conserved mechanism for the development of selective insulin resistance and thereby hyperglycemia.
Read the Publication in Elife (Open Access)
Abstract and figure from Shimobayash et al. (2023) Elife published under a CC BY 4.0 license.