How to undo splicing deficits
With the ever better understanding of regulated gene expression, it has become increasingly clear that RNA is much more than a passive intermediate. Amongst these transcription-translation regulation processes, splicing is a particularly crucial step – crucial, but also tricky. Splicing deficits could be associated with a wide range of maladies, so finding ways to deal with these deficits might open up new clinical paths with big therapeutic potential. The most promising candidates for this are small unusually structured drugs called oligonucleotides which have the power to shift the splicing process. If this shift becomes controllable we might dispose of a clinical tool to undo changes due to mutations in disease processes.
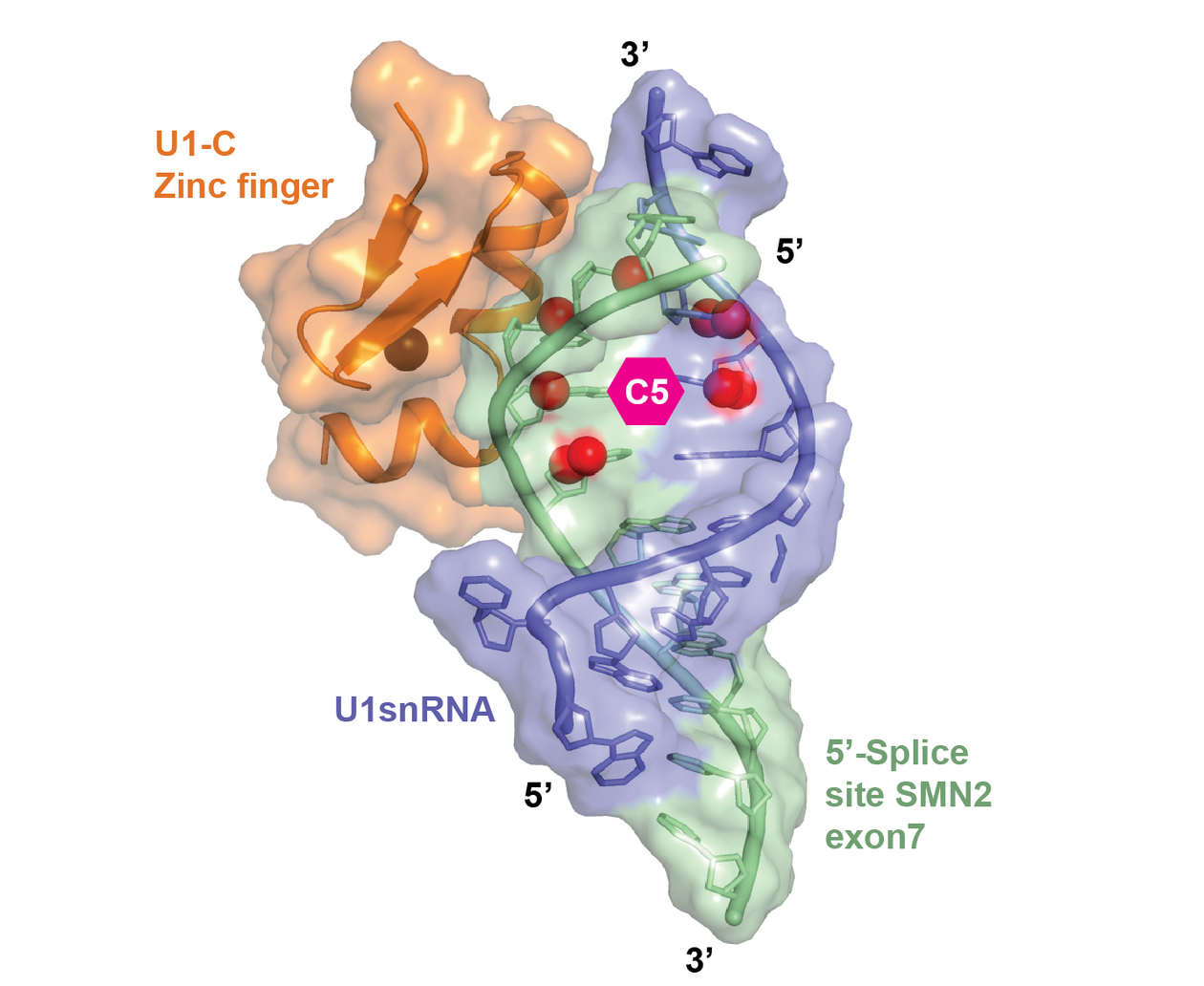
One of these diseases is spinal muscular atrophy (SMA), a rare genetic condition caused by the mutation or deletion of the survival of motor neuron 1 (SMN1) gene. SMA is characterized by progressive degeneration of spinal motoneurons. Humans carry two nearly identical copies of the SMN genes: SMN1 and SMN2. SMN1 expresses the full-length SMN protein, whereas SMN2 leads to a mRNA that is translated into a truncated and unstable protein. This is due to a C-to-T transition at position 6 of exon 7, causing exon 7 to be predominantly skipped. The nucleotide substitution in SMN2 results in the production of around 80 – 90% of truncated proteins and only 10 – 20% of functional full- length proteins.
In a recent Nature communications paper, a research group from Roche in collaboration with the Allain group from the Institute of Molecular Biology & Biophysics at ETH Zuerich reports a striking success in targeting these kind of deficits. Their report suggests that a splicing deficit of an individual gene can be selectively targeted through small molecule interactions with RNA:protein complexes. In the paper the researchers stress that as missplicing is the cause of many disease-causing mutations, the work has the potential to have widespread implications in the research and development of such RNA-targeting therapies.
Small molecule splicing modifiers have been previously described, but they targeted the general splicing machinery with a low specificity for individual genes. The group of molecules found and described by the joint group is much more promising for thera- peutic use. By using a combination of RNA splicing, transcription, and protein chemistry techniques, the researchers were able to show that these molecules directly bind to two distinct sites of the SMN2 pre-mRNA, thereby stabilizing a yet unidentified ribonucleoprotein complex that is critical to the specificity of these small molecules for SMN2 over other genes. They as well found that the molecules function via interaction with a tertiary RNA structure comprising the ESE2 region of exon 7, and an RNA helix that binds the U1 snRNP complex. This interaction of small molecules with the mRNA:protein complex is critical for the high selectivity of the small molecules for SMN2, which is higher than that of compounds that interact only at the 5'ss site.
In addition to the therapeutic potential of these molecules for treatment of SMA, the work has wide ranging implications in understanding how small molecules can interact with specific quaternary RNA structures. This is potentially relevant in cases of previously considered undruggable small molecule targets.
Sivaramakrishnan M. et al. (2017) Nature Communications 8(1), 1476 et al. (2017) (open access)
By Roland Fischer