CLIR sight on protein–RNA interactions
A new method to study protein–RNA interactions at amino acid and nucleotide resolution is proposed by Frédéric H.T. Allain, Ruedi Aebersold et al. It has been tested successfully on the complex of polypyrimidine tract binding protein 1 with a natural RNA target and is expected to be applicable to any RNP (of bigger size as well) for elucidating protein–RNA interactions and generating and refining precise structural models.
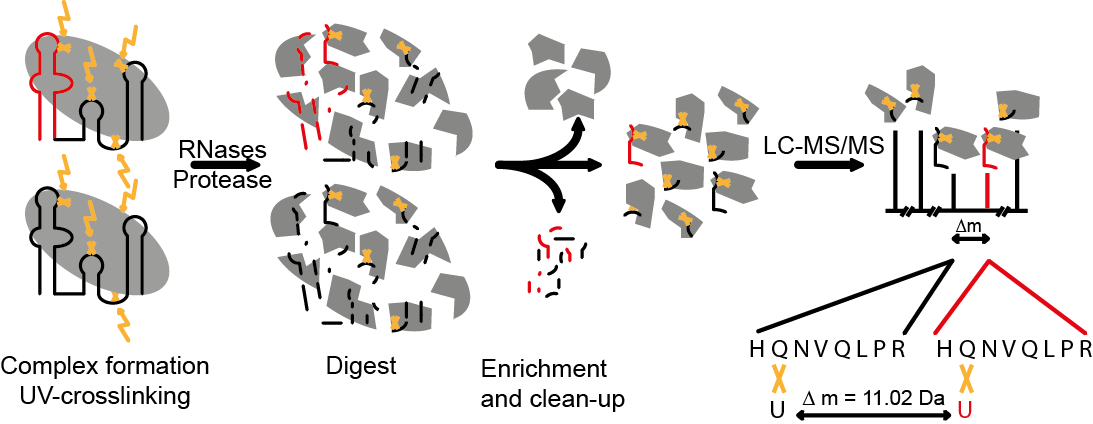
Ribonucleoproteins (RNPs) are key regulators of cellular functions such as gene expression. Even single nucleotide mutations can alter RNA–protein interactions with fatal consequences. Deciphering protein–RNA interactions at single amino acid and nucleotide resolution would therefore enable further functional characterization of RNPs, but until now, the exact positions of the proteins on the RNA have remained inaccessible. A group around Frédéric H.T. Allain from the ETH Institute of Molecular Biology and Ruedi Aebersold from the ETH Institute of Molecular Systems Biology and Biophysics led by Georg Dorn (Allain group ) and Alexander Leitner (Aebersold group) has recently presented an efficient approach, termed CLIR-MS/MS, to localize protein–RNA interactions simultaneously at amino acid and nucleotide resolution.
CLIR stands for "cross-linking of segmentally isotope labeled RNA". In combination with tandem mass spectrometric analysis (MS/MS), the CLIR labeling strategy uses the introduction of stable isotopes to locate the interaction site(s) between protein(s) and RNA. The group first applied the method to the complex of polypyrimidine tract binding protein 1 (PTBP1) with a natural RNA target. PTBP1 is a key alternative splicing factor and a major internal ribosomal entry site (IRES) trans-acting factor of several cellular and viral mRNAs. It was used as a model system because it is of biological relevance and has been studied in Fred Allain's group by NMR spectroscopy and other methods.
PTBP1 contains four RNA recognition motifs (RRM), whose structures in complex with a small single-stranded CUCUCU motif were determined by NMR spectroscopy. However, the recognition of guanines by PTBP1 and cooperative binding of all four RRMs to a large and structured RNA remain unexplained. The authors used MS and NMR spectroscopy to study PTBP1 in complex with a structured RNA molecule consisting of domains D–F of the IRES of encephalomyocarditis virus. This IRES part binds all four RRMs of PTBP1 and is essential for the regulatory function of PTBP1 in translation initiation.
Taking advantage of the ability of CLIR-MS/MS for high-resolution protein–RNA interaction mapping, the group used the identified crosslinks as intermolecular distance restraints (a form of spatial/distance information) for structural modelling, combined with restraints derived from available structural data of PTBP1-RRMs and from RNA structure predictions. In the context of CLIR-MS/MS, a certain amino acid and nucleotide must be close to each other in 3D space in order to be cross-linked. This "restraint" can be used in computational modelling methods to calculate the structure of protein-RNA complexes. Notably, all except one CLIR-MS/MS distance restraints were fulfilled by a single conformation for each RRM.
The authors believe that the method is applicable to any RNP of interest for elucidating protein–RNA interactions and generating and refining precise structural models of such RNPs. Furthermore the authors successfully tested the applicability of CLIR-MS/MS to larger RNPs as well – they expect that it can be used to study more complex systems such as in vitro reconstituted multicomponent RNPs. Fred Allain is convinced that the method is “a new approach that could considerably speed up the time needed to determine structures of protein-RNA complexes.”
Dorn G. et al. (2017) Nature Methods 14(5), 487-490
By Roland Fischer